Aktualności

„Przede wszystkim nie iść tą drogą samemu”. Rady dla naukowców, którzy chcą wejść w świat biznesu

Dr Alicja Mikołajczyk wytypowana jako Rising Star in Materials Science 2023
Rising Stars in Materials Science to wyróżnienie przyznane 17 początkującym naukowcom, którzy reprezentują rozległość, różnorodność i wzajemne powiązania nauk o materiałach prowadzonych na całym świecie. W grupie…

Na Międzyuczelnianym Wydziale Biotechnologii powstał kącik zabaw dla dzieci
Specjalnie wydzielona przestrzeń powstała w celu stworzenia przyjaznego i funkcjonalnego środowiska dla pracowników, doktorantów i studentów wydziału oraz ich dzieci. W minioną środę, 24 kwietnia, na Międzyuczelnianym…
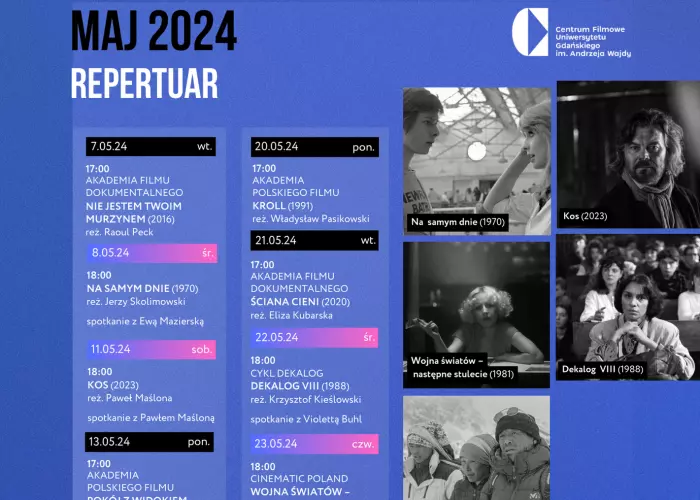
Maj w Centrum Filmowym UG im. A. Wajdy
Maj tuż tuż, więc zachęcamy do zapoznania się z repertuarem Centrum Filmowego UG im. Andrzeja Wajdy! Poniedziałkowe seanse spod znaku Akademii Polskiego Filmu to darmowe lekcje polskiego…

Prewencja osteoporozy. Rusza skierowany do kobiet program „Nauka dla Społeczeństwa II”
Osteoporoza to choroba charakteryzująca się obniżeniem masy kostnej, a także pogorszeniem jakości oraz zaburzeniami mikroarchitektury kości. W konsekwencji może to prowadzić do osłabienia…

Zgłoś swój pomysł na Majówkę z UG!
Centrum Aktywności Studenckiej i Doktoranckiej UG zaprasza do wzięcia udziału w organizacji „Majówki z UG”. Każdy może zgłosić swoją propozycję na aktywność, którą chciałby zrealizować…

Inspirująco podczas czwartego dnia Intellectual Property & Innovation Week
Czwarty dzień konferencji Intellectual Property & Innovation Week został poświęcony przedsiębiorczości. Studenci biorący udział w wydarzeniu dowiedzieli się między innymi, jak z pomysłu…
O Uniwersytecie

O Uczelni
Informacje ogólne oraz historyczne, o władzach i strukturze organizacji, zamówieniach publicznych a także przewodnik po UG – mapa oraz wirtualny spacer.

Nauka
Informacje o badaniach naukowych, projektach krajowych i zagranicznych, konferencjach, seminariach i wyjazdach zagranicznych, stypendiach, nagrodach, urlopach naukowych, a także stopniach naukowych i tytułach.

SEA-EU
SEA-EU to sojusz 9 europejskich i nadmorskich uczelni z Gdańska, Kadyksu, Brestu, Kilonii, Splitu, Malty, Algarve, Neapolu oraz Bodø. Naszą wizją jest stworzenie międzynarodowego i interdyscyplinarnego Uniwersytetu Europejskiego.

Rekrutacja
Zapraszamy na studia: I i II stopnia, jednolite magisterskie, podyplomowe, do szkół doktorskich. Tutaj również oferta kursów, szkoleń i szkół letnich.

Współpraca
Informacja o współpracy: międzynarodowej, międzyuczelnianej, z otoczeniem społeczno-gospodarczym, która realizowana jest w ramach projektów m.in. Erasmus+, Welcome to Poland (NAWA).

Erasmus+ InnHUB – finansowanie projektów
Międzysektorowe centrum innowacji integrujące potencjał uczelni, szkół, ośrodków kształcenia zawodowego, przedsiębiorców na rzecz budowania systemu edukacji i kształtowania kompetencji zawodowych.
0
Studentów
0
Dydaktyków
0
Wydziałów/ Międzynarodowe Agendy Badawcze
0









